
靶向蛋白质降解(TPD)是一种通过诱导E3泛素连接酶与靶向蛋白接近而实现的技术,这一过程有助于促进目标蛋白的泛素化及随后在蛋白酶体中的降解。作为一种新兴的治疗手段,TPD在处理致病蛋白方面展现出显著潜力。传统上,TPD通过两种方式实现:一是利用蛋白质降解-靶向嵌合体(PROTACs),这是一种双功能化合物,由两个独立部分组成,分别与靶点和E3连接酶相结合;另一种则是使用单价结合连接酶或靶点的分子胶。
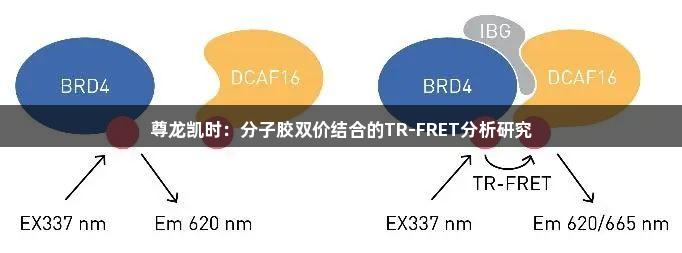
在本文中,我们将重点介绍一种称为分子内双价粘合剂(Intramolecular Bivalent Glues, IBGs)的新型BRD4降解剂及其作用机制,展现其为靶向蛋白质降解带来的全新模式。与PROTACs通过trans构象连接目标蛋白与E3连接酶不同,IBGs以cis构象同连接目标蛋白的两个相邻结构域,从而增强与E3连接酶的表面互补性。这种构象的改变利用了目标蛋白与连接酶之间的天然亲和力,使BRD4被“粘附”在E3连接酶DCAF16上,从而实现BRD4的降解,而在没有IBG的情况下,此过程不会发生。
通过对BRD4–IBG1–DCAF16三元复合物结构的解析,研究进一步指导了高效降解剂的理性设计,使其效能达到了低皮摩尔级别。我们采用时间分辨荧光共振能量转移(TR-FRET)法评估BRD4与DCAF16之间的三元复合物形成。该分析利用针对BRD4的铕标记抗体和Cy5标记的DCAF16。如果IBG分子能够诱导标记的BRD4与DCAF16之间形成三元复合物,荧光团之间的距离将足够近,形成荧光共振能量转移(FRET)现象。因此,激发铕供体荧光团将导致能量转移,Cy5受体荧光则可通过酶标仪进行测量。
我们在TR-FRET分析中,将Cy5标记的DCAF16、His-BRD4与铕标记抗His抗体的储备溶液均配制在TR-FRET缓冲液中(50mM HEPES,pH 7.5,100mM NaCl,1mM TCEP,0.05% Tween-20)。便进行了两类TR-FRET实验:(1)将IBGs梯度滴定加入BRD4与Cy5-DCAF16混合物中,分析复合物形成;(2)将Cy5-DCAF16滴定加入BRD4或BRD4+IBG中,分析复合物稳定性。
在我们的第一类实验中,将IBGs以1:4比例梯度稀释,加入至100nM BRD4与100nM Cy5-DCAF16的混合液中,置于白色384孔板中,每孔体积为16μL。对于复合物稳定性实验,Cy5-DCAF16同样以1:4比例稀释加入。最终BRD4的浓度为200nM,IBG1的浓度为1μM。无论哪种实验格式,铕标记抗His抗体和DMSO的浓度分别保持在2nM和0.5%。经过50g的离心处理后,反应板静置于室温30分钟,随后使用PHERAstar FS读板仪读取结果。
我们的研究目标是体外表征DCAF16、BRD4与双价分子胶IBG1之间可能的相互作用。通过等温滴定量热法(ITC)观察到IBG1、DCAF16与BRD4 Tandem(包含BD1和BD2两个溴结构域并由天然连接肽连接)形成三元复合物。TR-FRET复合物形成分析显示,在IBG1的滴定下,BRD4 Tandem与DCAF16间以剂量依赖的方式形成三元复合物(EC50=44nM)。总体而言,IBG1的活性相较于其单价前体JQ1更为卓越。
补充的TR-FRET稳定性分析进一步证实,IBG1存在时,滴定DCAF16至BRD4 Tandem时形成复合物(Kd=712nM)。令人惊讶的是,即使不添加IBG1,也可检测到DCAF16与BRD4 Tandem之间内在的亲和力(Kd=1μM),而这种亲和力在孤立的BRD4 BD1或BD4 BD2结构域中并未观察到,这表明两者之间需要协同作用才能形成复合物。对比分析显示,在IBG1存在下,DCAF16与BRD4 Tandem之间的亲和力显著增强(Kd由4μM降低至0.6μM),同时相互作用的热力学特性也发生改变。
在确认IBG1的作用机制后,研究人员合成了新的化合物IBG3,以增强其对于BRD4和DCAF16的粘合活性。新化合物IBG3在BRD4的降解效率方面优于IBG1,降解效率达到低皮摩尔水平(DC50=67pM)。在TR-FRET实验中,IBG3对BRD4–DCAF16复合体的粘合能力提升至(EC50=32nM)。与其前体化合物相比,IBG3对于BRD2和BRD4展现出更高特异性,更倾向于作用于串联溴结构域而非孤立结构域,并且其机制依然通过DCAF16介导,表明其仍然通过相同的分子内“胶合作用”实现降解。
综上所述,通过本文描述的TR-FRET相互作用分析方法,能够表征BRD4-DCAF16之间的新型分子胶。这类新型分子内双价胶通过同时结合目标蛋白的两个结构域,增强与E3连接酶的亲和力,从而形成新的相互作用模式。这一策略为靶向多种效应蛋白、重构细胞信号通路、实现蛋白降解等应用提供了广阔的药理学路径。欢迎关注尊龙凯时的官方平台,获取更多相关信息与研究进展。
